
Documentation of the GEMtractor
GEMtractor was published in GEMtractor: extracting views into genome-scale metabolic models at Oxford Bioinformatics. If you found GEMtractor useful for your analyses, please consider referencing it in your publications.
- What's the difference between enzyme-centric and reaction-centric?
- Does it make sense to extract the enzyme-centric network?
- Which type of models does the GEMtractor support?
- How does the GEMtractor identify enzymes that are associated to a reaction?
- Why do enzymes have such a strange name?
- What if no enzyme is associated to a reaction?
- What exactly happens when I remove a species during filtering?
- Why is a certain species highlighted in red?
- What exactly happens when I remove a reaction during filtering?
- Why is a certain reaction highlighted in red?
- What exactly happens when I remove an enzyme during filtering?
- Why is a certain enzyme highlighted in red?
- What exactly happens when I remove an enzyme complex during filtering?
- Why is a certain enzyme complex highlighted in red?
- What is an enzyme complex?
- How does the batch filtering work?
- How should I format my flux balance results?
- Why does it take so long?
- What are currency metabolites?
- How can I discard currency metabolites (such as H+, H2O or ATP)?
- How does the GEMtractor select the models from Biomodels?
- How does the GEMtractor select the models from BiGG?
- Which data does the GEMtractor store on the server?
- Why am I always redirected back to model selection?
- What are the different export options?
- Why is my exported model empty?
- Aren't the directions wrong?
- Enzyme or Gene? Species or Metabolite?
- How can I clear my data?
- Can I look inside the GEMtractor?
- How can I modify the default settings?
Let's have a look at the following Metabolite-Reaction Network, which is a tripartite graph consisting of tree types of nodes: Six metabolites (A, B, C, D, E, and F) interact in four reactions (r, s, t, and u) that are catalyzed by six enzymes or enzyme complexes (L, M, N, P+O, Q, and Q+R):
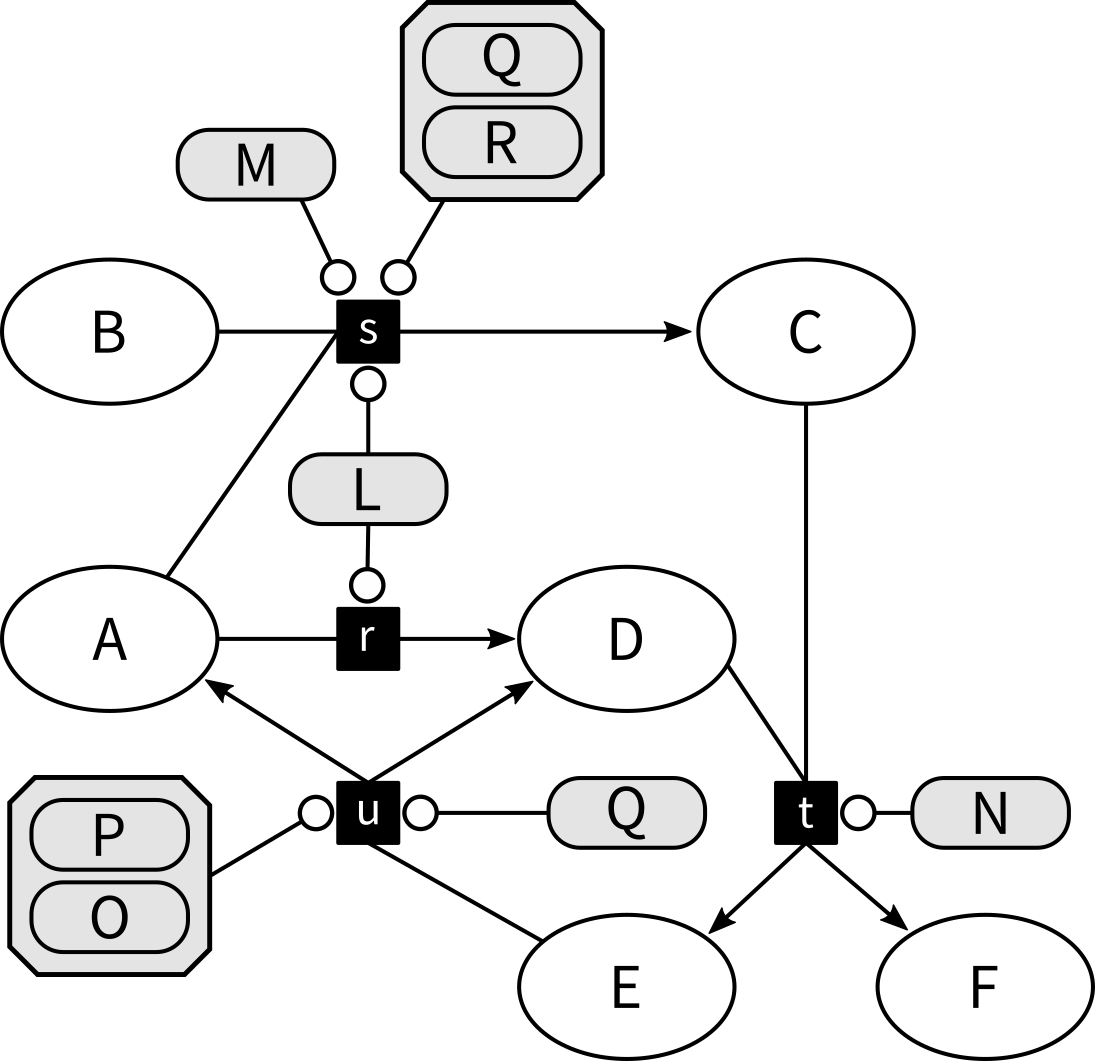
Here for example, the reaction r transforms A into D and this process is catalyzed by the enzyme L.
Similarly, s consumes A and B to produce C.
This reaction s is catalyzed by L or M or Q+R.
From this Metabolite-Reaction Network we can extract the Reaction-centric Network, which is a unipartite graph — all its nodes are of the same type:

Here, an arrow between two reactions indicates, that there is at least one species which is produced by the source of the arrow and consumed by the target of the arrow.
For example, s is linked to t, because s produces a metabolite (C) that is consumed by t.
And similarly, every reaction that produces D (r and u) is linked to every reaction that consumes D (t), resulting in the two edges r→t and u→t.
Furthermore, we can extract the Enzyme-centric Network, which is again a unipartite graph as its nodes represent enzymes:
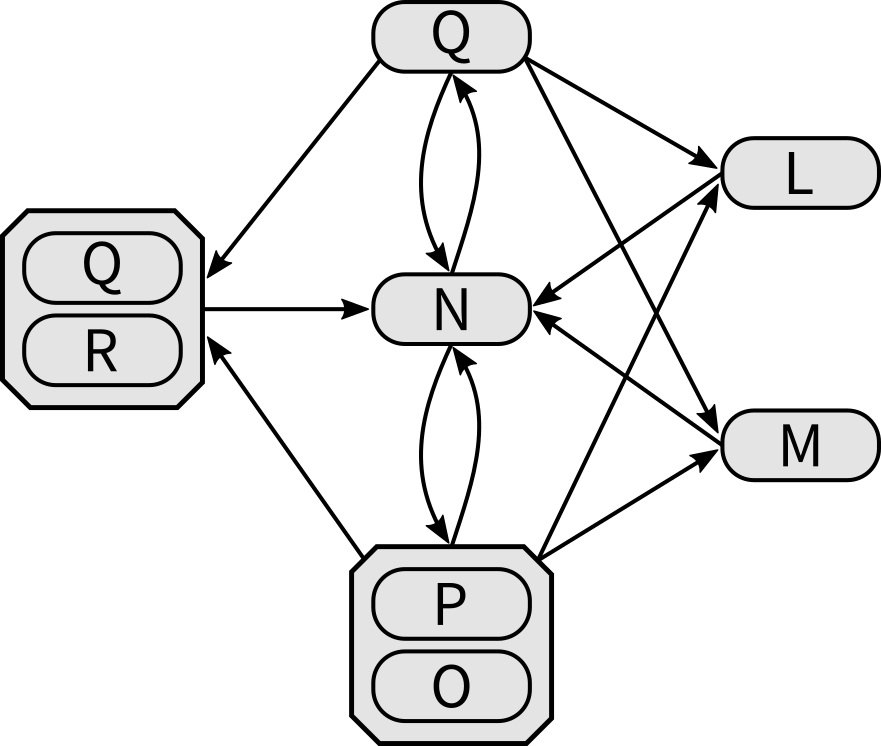
Based on the Reaction-centric Network every node and its edges is cloned according to the alternative enzymes that catalyze the corresponding reaction.
For example, s is linked from u and is linked to t.
As s is catalyzed by L or M or Q+R, it is unfolded into three nodes, each receiving a link from u and a link to t.
The reaction t is catalyzed by N exclusively, so it's basically just replaced.
Eventually, we'll find a link from each of L, M, and Q+R to N.
Did you notice, that the topology of the Enzyme-centric Network differs a lot from the topology of the Reaction-centric Network? We think it's worth analyzing that :)
A Metabolite-Reaction Network typically encodes for a complex graphs of species, reactions, and enzymes. Thus, comparing genome-scale models through topological analysis of such a multipartite graph is challenging. However, in many practical cases it is not necessary to compare the full networks. Using the GEMtractor you can trim models, for example according to the results of other methods such as flux balance analyses, which identify subnetworks from a whole genome metabolic model.
The Enzyme-centric Network is then a simplified presentation of the metabolic network at the protein scale. All nodes in that network are of the same type, facilitating topological analyses. In fact, the enzyme-centric networks provide us a critical precursor of physiological representation from genomics data, by determining metabolic distances between enzymes.
The GEMtractor supports models encoded using the Systems Biology Markup Language (SBML) format. SBML is a standard format for computer models of biological processes. You can find many example models at Biomodels or at BiGG Models.
Enzymes are based on genes, they are the gene's products. The GEMtractor currently supports two formats of gene associations in SBML documents: The legacy SBML-notes annotations, and well-structured annotations using the FBC package in SBML Level 3 documents.
An example of legacy SBML-notes annotations from MODEL1011080003, which encodes for the gene combination
438 or 330:
<reaction id="R_METSR_S1" [...]> <notes> <html:p>GENE_ASSOCIATION: ( 438 or 330 )</html:p> <html:p>PROTEIN_ASSOCIATION: MsrA</html:p> <html:p>SUBSYSTEM: S_Amino_Acid_Metabolism</html:p> <html:p>PROTEIN_CLASS:</html:p> </notes> [...] </reaction>
An example of a well-structured gene associations from e_coli_core using FBC-annotations, which encodes for the gene combination
(G_b3738 and G_b3736 and G_b3737 and G_b3735 and G_b3733 and G_b3731 and G_b3732 and G_b3734) or (G_b3734 and G_b3732 and G_b3731 and G_b3733 and G_b3735 and G_b3737 and G_b3736 and G_b3738 and G_b3739):
<reaction id="R_ATPS4r" [...]> [...] <fbc:geneProductAssociation xmlns:fbc="http://www.sbml.org/sbml/level3/version1/fbc/version2"> <fbc:or sboTerm="SBO:0000174"> <fbc:and sboTerm="SBO:0000173"> <fbc:geneProductRef fbc:geneProduct="G_b3738" /> <fbc:geneProductRef fbc:geneProduct="G_b3736" /> <fbc:geneProductRef fbc:geneProduct="G_b3737" /> <fbc:geneProductRef fbc:geneProduct="G_b3735" /> <fbc:geneProductRef fbc:geneProduct="G_b3733" /> <fbc:geneProductRef fbc:geneProduct="G_b3731" /> <fbc:geneProductRef fbc:geneProduct="G_b3732" /> <fbc:geneProductRef fbc:geneProduct="G_b3734" /> </fbc:and> <fbc:and sboTerm="SBO:0000173"> <fbc:geneProductRef fbc:geneProduct="G_b3734" /> <fbc:geneProductRef fbc:geneProduct="G_b3732" /> <fbc:geneProductRef fbc:geneProduct="G_b3731" /> <fbc:geneProductRef fbc:geneProduct="G_b3733" /> <fbc:geneProductRef fbc:geneProduct="G_b3735" /> <fbc:geneProductRef fbc:geneProduct="G_b3737" /> <fbc:geneProductRef fbc:geneProduct="G_b3736" /> <fbc:geneProductRef fbc:geneProduct="G_b3738" /> <fbc:geneProductRef fbc:geneProduct="G_b3739" /> </fbc:and> </fbc:or> </fbc:geneProductAssociation> [...] </reaction>
The GEMtractor then uses the identifiers of the gene-products to refer to enzymes.
Gene products that are combined using a logical or are assumed to be alternatives for a catalysis.
Gene products that are combined using a logical and are assumed to build an enzyme complex, in which all enzymes need to be present for a catalysis. Thus, a geneProductAssociation encoding for a and (b or c) means, that the reaction is catalysed by a and b or a and c.
The enzyme names used by the GEMtractor are actually the gene-products' identifiers as stated in the SBML document.
Those are typically generated by some kind of software and often just numbered with a optional prefix (such as g1, g2, g3, or b0116 etc).
To get more information about a certain enzyme, you need to look into the model's SBML code.
If you're lucky, the gene product is properly annotated and you'll find links to other resources describing the enzyme.
See also What if no enzyme is associated to a reaction?
If the GEMtractor cannot find a gene-association, it assumes there is only a single gene and assigns it the identifier of the reaction prefixed with "reaction_".
Thus, you can easily spot fake enzymes.
When exporting the model, you may also discard these invented enzymes.
See also How does the GEMtractor identify enzymes that are associated to a reaction?
Trimming the species of a model actually means removing species from reactions. That is, trimmed species will not appear in any reaction anymore, but if you export the Metabolite-Reaction Network, they may still appear in the model (as they may still be required by other elements in the model). We call these species ghost species. When exporting your model, you will find an option to entirely remove such ghost species from the model. See also What are the different export options?
If you remove a species from a network, all the reactions that consume or produce this species may now be invalid. Thus, the reactions relying on a removed species will be highlighted in red in the Reactions tab, as you may want to re-evaluate these reactions.
A species is highlighted in red if you trimmed all reaction in which it appeared. So you may want to re-evaluate your filters or trim that species as well. In that case, a small info-icon will appear behind the species' identifier and you can move your mouse over that icon to get more information.
A trimmed reaction will disappear from the original model prior to subsequent conversions. All species and enzymes linked to that reaction (as reactants, products, or gene-association) will be unlinked. Thus, there may be species, enzymes and enzyme complexes, which do not appear in the model anymore. Those left-overs will be highlighted in red to raise your attention, as you may want to re-evaluate your filters.
There are two reasons why a reaction will be highlighted in red:
- One of the species it consumes or produced is trimmed from the model — so the reaction may be invalid now.
- All the enzymes or enzyme complexes required to catalyze this reactions are trimmed from the model, thus the reaction probably cannot take place anymore.
If you trim an enzyme from your model, this enzyme will be removed from all reactions that it catalyzes. This may result in the following inconsistencies:
- A reaction looses all its enzymes and enzyme complexes (and probably won't take place anymore).
- The enzyme is part of an enzyme complex — thus, you may also need to remove the complex.
When exporting the model, you have the option to remove all reactions without enzymes and to remove complexes that lack an enzyme. See also What are the different export options?
An enzyme is highlighted in red, if your filters removed all reactions and enzyme complexes in which this enzyme participates. Thus, it is not appearing in the model anymore and you may want to trim it as well. A small info-icon will appear behind the enzyme's identifier and you can move your mouse over that icon to get more information about the reason for the inconsistency.
If you trim an enzyme complex, it will be removed from all reactions that it catalyzes. This may result in the following inconsistencies:
- A reaction looses all its enzymes and enzyme complexes (and probably won't take place anymore).
- An enzyme does not participate in any reaction or enzyme-complex anymore.
The inconsistent reactions or enzymes will be highlighted in red.
In addition, there may be an enzyme-complex superior to a trimmed complex — the trimmed enzyme complex consists of a set of enzymes, which is a subset of another enzyme complex.
Let's for example say you remove the enzyme complex A+B and there is another enzyme complex A+B+C.
It is very likely, that A+B+C is not possible if A+B is not possible.
However, we can think of inverse situations.
Thus, enzyme complexes are highlighted in red-italics, if any sub-complexes are trimmed.
So, if you remove A+B, then A+B+C will become red-italics.
When exporting the model, you have the option to remove all reactions without enzymes and to remove complexes that lack an enzyme. See also What are the different export options?
An enzyme complex will be highlighted in red, if it doesn't appear in any reaction anymore, or if one of its enzymes was trimmed.
The enzyme complex will be highlighted in red-italics, if an sub-complex was trimmed.
For example, if you remove A+B, then A+B+C will become red-italics.
A small info-icon will appear behind the enzyme's identifier and you can move your mouse over that icon to get more information about the reason for the inconsistency.
An enzyme complex is an enzyme, which consists of a complex of gene products.
So there may be two gene products A and B forming an enzyme complex A+B.
Nevertheless, A and B may as well catalyze reactions as individual enzymes.
We implemented batch filtering to support you in automatic filtering without implementing an API client. If you for example have a list of species, that you want to exclude according to your FBA result, you can just dump this list as comma-separated values and copy it to the batch text-field. Thus, you may save a lot of clicks compared to deselecting every species individually.
The text-field expects four lines, each starting with either species, reactions, enzymes, or enzyme_complexes, followed by a color (:) and the list of comma-separated entity-identifiers.
Applying the batch-filter will send the lists to the backend, where they will stored in your session, and reload the page to update the trimming selection.
GEMtractor allows for integrating the results of flux balance analysis. If you provide the fluxes of your model, another column in the table for filtering reaction will appear that lets you sort the reactions by their fluxes. To upload your fluxes you can use the upload box in the Reactions tab of the Trim page. Your fluxes need to be encoded in CSV, each row containing one reaction identifier and the corresponding flux (or vice versa: flux,reaction_id).
As GEMs are typically pretty big it takes some time to process them. We have of course been aware of that when developing the GEMtractor and implemented the modules with a strong focus on efficiency. And it actually works quite well for most models, but some models cause a significant delay. Nevertheless, let's have a look into the different delays to give you a feeling about what you're waiting for. We distinguish the following waiting times:
- When selecting a model: (i) If you select a model from BiGG or Biomodels, the GEMtractor need to download the model. Some of those models are extremely big, with up to 200 MB in size! Thus, it takes some time to obtain the model's SBML code — the model's name in the table is replaced by a loading indicator to let you know that something's happening. (ii) If you upload a model, it may also take some time depending on the size of your model and your internet connection. If this delay is a problem for you, you may want to run your own GEMtractor locally to get rid of the upload-waiting-time.
-
When clicking Trim: To start the trimming process, your browser needs the necessary information about the network encoded in your model.
Thus, at the server-side the GEMtractor needs to (i) read the model, (ii) extract the network, (iii) unfold the gene-association, (iv) serialize all the information in JSON and send it to your browser.
Typically, all sub-tasks grow with the model size, and so grows the waiting time.
However, some models come with pretty complex gene-association, such as
(A or B) and (C or D or E) and (F or G or H), which unfolds into 18 possible enzyme combinations... This increases the computational time exponentially. That's all done on our servers which are already quite fast, so there is not much you can do about it. Unless you have a faster server — then it would make sense to run your own GEMtractor. - While Trimming: As soon as you obtained the model's network, you can start trimming the model. The whole trimming process is done in your browser — thus, the performance depends on your local computer/laptop. For most models you won't recognize a delay, but for some models it will take a second or two after you (de)select a model entity, because your browser needs to recompute the trimmed whole network to check for inconsistencies. If you for example remove a reaction, it needs to check all species, enzymes, and enzyme-complexes to see if they now lack a connection in the model. Thus, in case of Recon3D it would need to re-evaluate almost 25 thousand model entities! And that may take a second on old laptops or out-dated browsers. Change/upgrade your browser or switch to a faster computer to speed-up this process.
- When Exporting: Exporting the trimmed model takes about as long as waiting for the network when clicking Trim, because the task is similar and it's all done on the server-side: The GEMtractor needs to (i) read the model, (ii) apply your trimming filters, (iii) extract the network, (iv) unfold the gene-association, and (v) serialize the network in your chosen format. The serialization process is fast and thus neglectable. The waiting time may however decrease a lot if you trimmed a lot — if there are for instance just a few reactions left, there won't be much to extract or unfold... Thus, to improve the speed you can trim more entities or deploy the GEMtractor on a faster server.
Currency metabolites are species in a model, which are used in almost every circumstance, as a medium of exchange. They are typically the most abundant species, so that their concentrations put no constraints on the activity of a reaction — they can be regarded as externally buffered with respect to the system [Huss and Holme2006].
See also How can I discard currency metabolites (such as H+, H2O or ATP)?
The tables listing the entities at the trimming page are sortable. Thus, you can click the header cell of a column to sort it ascending — if you click it again, the column will be sorted descending.
If you want to discard currency metabolites, you can sort the species table by the occurences.
This will bring the currency metabolites to the top of the page, making it easy to get rid of them, as shown in this screen shot:
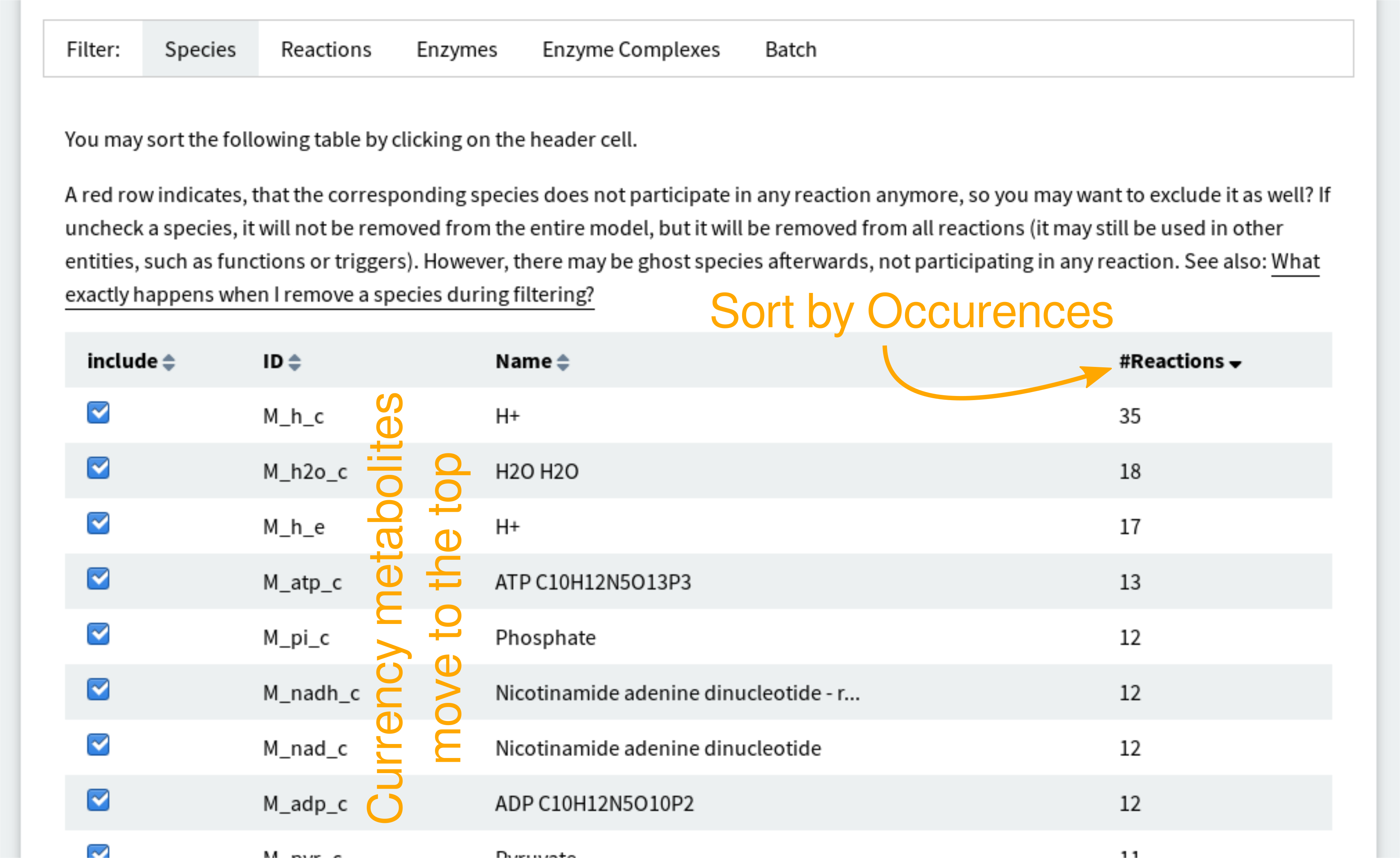 See also What are currency metabolites?
See also What are currency metabolites?
Biomodels is a high-quality resource for GEMs encoded in SBML. They actually feature thousands of genome-scale metabolic models! However, some of them are not useful or useable in GEMtractor (e.g. because of model encoding issues or missing gene associations). Thus, we decided to maintain a curated list of useable models. This list is pretty arbitrary - just drop us an email if you're missing a model in that list.
BiGG Models is a high-quality resource for GEMs encoded in SBML. They currently provide more than hundred SBML models including FBC annotations (see How does the GEMtractor identify enzymes that are associated to a reaction?). The table at the model-selection tab basically lists all available models from BiGG Models.
If accessed through a webserver, it stores the uploaded model (or the model downloaded from BiGG or Biomodels) and some session related settings. To see what the GEMtractor stores about you and how to delete the data it stores, head over to the privacy policy.
You probably do not accept cookies..? The GEMtractor uses sessions to store your settings. These sessions rely on a cookie in your browser. If the GEMtractor doesn't know which model you selected, it won't let you do for filtering or exporting... So you'll always be redirected to model selection.
To learn more on what's stored in your session have a look into the privacy policy.
When you're done trimming the model, you can export the resulting network in three different flavors:
- The Enzyme-centric Network is a unipartite graph. Every node represents an enzyme and every edge indicates, that the source of the edge catalyzes a reactions, which produces a metabolite that is consumed by another reaction catalyzed by the target of the edge.
- The Reaction-centric Network is a unipartite graph of reactions and their interactions. Two reactions are linked, if the source produced a metabolite that is consumed by the target of the edge.
- The Metabolite-Reaction Network is a tripartite graph, that includes metabolites, reactions, and enzymes and their interactions.
You can export each flavor in different formats:
- SBML — a standard format for computer models of biological processes. SBML is the only format that allows for proper annotations of entities. By design, SBML is bipartite. Thus, for the unipartite networks (Enzyme-centric and Reaction-centric) the GEMtractor needs to invent fake-reactions to link two nodes. These fake reactions have exactly one input and one output. However, if it's mandatory to receive a unipartite network and you don't need the annotation, you should go for one of the other formats. See also sbml.org
- GraphML — a graph format based on XML. In addition to the core network descriptions, the GEMtractor attaches a shape, a label, and a type to the nodes in the nodes in the graph. See also
- DOT — a graph description language with a simple syntax and many converters and tool support. See also Wikipedia:DOT
- GML — a graph description language with a simple syntax. See also Wikipedia:GML
- CSV — comma separated values. This just lists the edges' sources and targets, separated with a comma :)
A few more export options provide control over inconsistencies in the trimmed graph:
- Remove reactions, whose enzymes were all removed: You may have trimmed all the enzymes and enzyme complexes that catalyzes a certain reaction, but didn't trim the reaction itself — so the reaction is still in the mode, but it may not occur anymore. Should such reactions without enzymes be removed as well? (If you export the Enzyme-centric Network removing these reactions is mandatory: As there are no enzymes there is no Enzyme-centric Network to extract..)
- Remove ghost species: After trimming there may be species in the model, that do not appear in any reaction anymore — we call those ghost species. They may still be required in other entities in the model, that's the GEMtractor keeps them in the model by default. Do you still want to remove those species? This decision only affects the Metabolite-Reaction Network.
- Discard fake enzymes: When a reaction does not have an annotation with a gene-association. In these cases, the GEMtractor invents an enzyme (see What if no enzyme is associated to a reaction?). Should these fake enzymes be removed prior to subsequent analyzes?
- Remove reactions, in which at least one species was removed: If you trim a species off a reaction, this reaction (and its kinetic laws) may become invalid. Should reactions, which do not have all original species anymore, be removed?
- Removing an enzyme removes all complexes in which it participates: A trimmed enzyme may be part of an enzyme complex — thus, the enzyme complex may be invalid without a certain enzyme. However, it could as well be, that the enzyme's active site is broken for a certain substrate, but is still fully functional in a complex with other enzymes... So it's up to you: Should enzyme complexes be removed if an individual enzyme is trimmed?
Please note, that it may take a few seconds (sometimes minutes) to generate the different output files, depending on the size of your model. So don't get nervous — the click will return! (Sometimes even with an error ;-)
If you are working on a model, which does not define gene-associations, the GEMtractor invents fake enzymes (see What if no enzyme is associated to a reaction?). Consequently, if you want to export the enzyme-centric network of such a model, but choose to discard fake enzymes (see What are the different export options?), you will end up with an empty skeleton of your export format (e.g. an SBML model without species or reactions).
Maybe... And if you think we did a mistake please do not hesitate to contact us!
However, there are some sommon pitfalls.
Let's for example have a look at the following Metabolite-Reaction Network
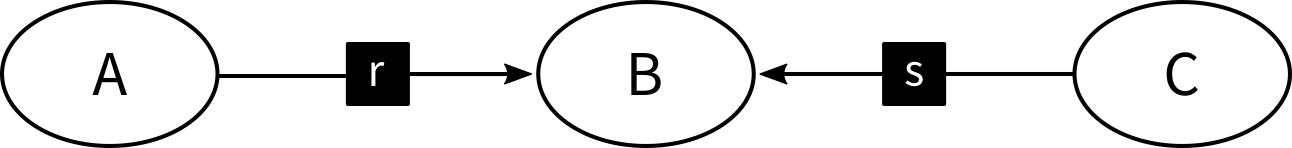 which encodes for a reaction
which encodes for a reaction r that consumes A and produces B, and another reaction s that consumes C and produces B.
Given this network, you could anticipate, that the reaction-centric network contains no edges.
However, the SBML code may define s to be reversible!
This may only be defined implicitely: In SBML prior to level 3 all reactions are in fact reversible, unless explicitly defined otherwise.
Thus, the network encoded in the model actually looks like this:
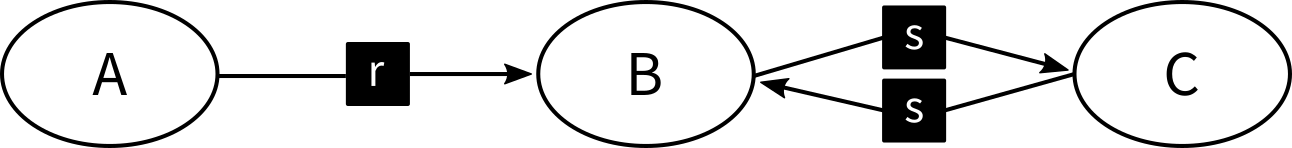 And suddenly, there is an edge in the reaction-centric network:
And suddenly, there is an edge in the reaction-centric network:
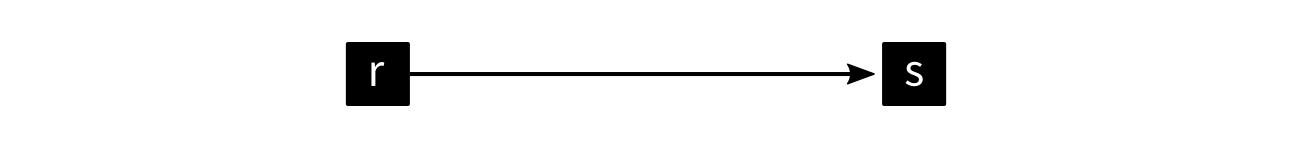 The same also applies to enzyme-centric networks.
The same also applies to enzyme-centric networks.
Yes, the wording is sometimes difficult. We tried to homogenize everything, but at some points it's inconsistent. For example:
- A reaction is basically catalyzed by an enzyme. In SBML, however, those enzymes are called gene-products. And gene-products are direct results from genes. Depending on the context, we use these terms as synonyms, but try to stick with enzyme.
- While GEMs are typically about metabolites interacting in reactions, in SBML they are more generically referred to as species. Even though, the GEMtractor is focused on GEMs, we basically also support all kinds of models, as long as they are implementable in SBML. For example, you can also trim tiny kinetic models! Thus, we prefer to use the term species.
- Is it an Enzyme-Centric Network or an Enzyme-Enzyme Network or and Enzyme-Interaction Network? People use seem to use different terms to refer to the same thing: The network of enzymes and their phenotypical interactions. We decided to call it Enzyme-Centric Network (for the enzyme interactions) and Reaction-Centric Network (for reaction interactions).
- Do you trim a model? Or reduce it? Or cut it? Or filter entities? All these phrases are synonyms to us. However, for the GEMtractor we prefer to talk about trimming of models :)
To see what the GEMtractor stores about you and how to delete the data it stores head over to the privacy policy.
Sure - the GEMtractor is free software! :)
We develop the source code at an internal repository, but automatically push updates to the GitHub at binfalse:GEMtractor. Documentation for the code is available from doc.bio.informatik.uni-rostock.de/GEMtractor. We are looking forward for your code contributions, bug reports, and feature requests :)
The settings of the GEMtractor are controlled through environment variables on the server. Thus, you can only change the settings, when running your own GEMtractor server! Just scroll down to learn how to run your own GEMtractor — it's pretty easy.
Send Job as JSON
The API is listening at DOMAIN.URL/api/execute — it expects a job encoded as JSON and sent via HTTP POST, such as this:
{
"export": {
"network_type":"mn",
"network_format":"sbml",
"remove_reaction_enzymes_removed": False,
"remove_ghost_species": False,
"discard_fake_enzymes": False,
"remove_reaction_missing_species": True,
"removing_enzyme_removes_complex": True
},
"filter": {
"species": ["h2o", "atp"],
"reactions": [],
"enzymes": ["gene_abc"],
"enzyme_complexes": ["a + b + c", "x + Y", "b_098 + r_abc"],
},
"file": "<sbml>...</sbml>"
}
Keys of the job's JSON object
export
The key export is required, and it must encode for a JSON object containing the following keys:
network_type: String"en"or"mn"— do you just want the filtered metabolic network ("mn"), or should the GEMtractor return the extracted enzyme network ("en")?network_format: String"sbml","graphml","gml", or"dot"— how should the returned network be encoded?remove_reaction_enzymes_removed: Boolean — remove reactions, whose enzymes were all removed? (Optional, defaults totrue)remove_ghost_species: Boolean — remove species, which do not participate in any reaction anymore? (Optional, defaults tofalse)discard_fake_enzymes: Boolean — remove invented enzymes? See What if no enzyme is associated to a reaction? (Optional, defaults tofalse)remove_reaction_missing_species: Boolean — remove reactions, in which at least one species was removed? That is, if you remove a species from a reaction, should the GEMtractor also remove the whole reaction? (Optional, defaults tofalse)removing_enzyme_removes_complex: Boolean — remove enzyme complex, if one of its enzymes was removed? In other words: If you remove an enzyme, should all the complexes in which it participates also been removed? (Optional, defaults totrue)
filter
The key filter is optional.
You just need to provide it, if you want to get rid of some items in the model.
If provided, it may contain the following keys:
species: List of species-identifiers to remove from reactions.reactions: List of reaction-identifiers to remove from the model.enzymes: List of gene-identifiers to remove from reactions.enzyme_complexes: List of enzyme-complex-identifiers to remove from reactions. Every item must contain a string of enzymes in the corresponing enzyme complex joined with the string" + "code>. Thus, if you want to remove the complex formed by the enzymesA23andB_42, you would add"A23 + B_42"to this list.
id attribute of the SBML entity.
However, every list can also be empty.
file
The key file is required.
It must contain the raw SBML code of your input model, including the <?xml version="1.0" encoding="UTF-8"?> prefix.
Expect a response
Reading, filtering, converting, and exporting may of course take some time, but if the GEMtractor finishes successfullty it will return with an HTTP response status 200 and immeditaly serve the exported content.
If the GEMtractor was not successful, you'll get an HTTP status other than 200, for example:
400: Your request was bad, for example the JSON was not well formed or is missing a required key, or the supplied model is not valid SBML.302: You forgot to send the job as POST data. You'll, thus, be redirected to this section to lear how to fire a job :)500: There was a problem on the server, e.g. the GEMtractor could not generate the desired file.
Use in your application
It should be possible to integrate the GEMtractor in any application. Some example clients for different languages are supplied with the source code. Just have a look into the GitHub repository at binfalse:GEMtractor/clients.
GEMtractor comes with proper support for Docker. Thus, installing an own instance is pretty easy, including all benefits as increased speed and privacy.
We recommend doing so using Docker.
Just get a copy of the sources from GitHub:GEMtractor and you will find a Dockerfile and a docker-compose.yml file (see Docker Compose) in the root of GEMtractor's directory.
It should be sufficient, to just run
docker-compose up
and everything will be built and started. You'll then find the GEMtractor listening at port 80 on your local machine. Just open your web browser and browse to http://localhost. That's been easy, wasn't it? :)
Look inside the docker-compose.yml and check the Settings section below to configure your GEMtractor instance.
Those who want to deploy it to some local server will know what to do..
A quick node on the infrastructure: There are actually two services necessary to run the GEMtractor:
- A webserver to deliver static files and forward dynamic requests to the application server (we prefer using an Nginx)
- An application server to run the Python code (we prefer gunicorn)
Dockerfile and a docker-compose.yml file and check the nginx.conf.
However, all this setup is just our recommendation — you can of course use any other web server (e.g. Apache's httpd) or other means to server the Python parts (e.g. uwsgi)
There are a few environment variables that gives you some control about the behavior of the GEMtractor:
DJANGO_DEBUG: BooleanTrueorFalse— should Django run in debug mode? Do not set to 'True' in production! (default:True, currently:False)DJANGO_LOG_LEVEL: StringDEBUG,INFO,WARNING,ERROR, orCRITICAL— what kind of log messages should be reported? (default:True, currently:INFO)DJANGO_ALLOWED_HOSTS: String — additional hostname for which GEMtractor should be listening. (currently:['localhost', '127.0.0.1', 'gemtractor-py'])MAX_ENTITIES_FILTER: Int — up to how many entities in a model it should allow for filtering in the web browser? Web browsers will die if there are too many entities... (default:100000, currently:10000.0)STORAGE_DIR: Path — where to store models and exports etc. (default:/tmp/gemtractor-storage/, currently:/storage)KEEP_UPLOADED: Time in seconds — how long to keep uploaded files on the server? Everytime an uploaded file is used for filtering or exporting, the age of the file is reset to zero. (default:1.5*60*60= 1.5 hours, currently:5400.0seconds)KEEP_GENERATED: Time in seconds — how long to keep generated files on the server? Generated files are basically only for immediate delivering. (default:10*60= 10 minutes, currently:600.0seconds)CACHE_BIGG: Time in seconds — how long to cache the list of models available from BiGG Models? (default:60*60*24= 24 hours, currently:86400.0seconds)CACHE_BIGG_MODEL: Time in seconds — how long to cache a single model from BiGG Models? (default:7*60*60*24= 7 days, currently:604800.0seconds)CACHE_BIOMODELS: Time in seconds — how long to cache the search results of models available from Biomodels? (default:60*60*24= 24 hours, currently:86400.0seconds)CACHE_BIOMODELS_MODEL: Time in seconds — how long to cache a single model from Biomodels? (default:7*60*60*24= 7 days, currently:432000.0seconds)HEALTH_SECRET: Some secret that needs to be sent as JSON POST to/api/statusto get health information about the instance. If not explicitly set, everyone can get the health data.
export CACHE_BIGG=60 export MAX_ENTITIES_FILTER=20000 # [...] gunicorn --timeout 600 --bind 0.0.0.0:80 gemtractor.wsgi:applicatio
Thus, the running GEMtractor process will then cache the BiGG Models list for 60 seconds and allow for filtering of models in the browser with up to 20K entities.
When using Docker Compose, you would set the variables in the demo docker-compose.yml file, for example:
version: '3'
services:
gemtractor-py:
[...]
environment:
CACHE_BIGG: 60
MAX_ENTITIES_FILTER: 20000
[...]
GEMtractor is able to export some health data about the instance.
Just send a JSON request through HTTP POST to /api/status to learn how many files are cached, how many files are uploaded, how how much storage space is used.
You may protect this data by setting HEALTH_SECRET to something non-empty.
In that case, you need to submit the secret in the JSON POST object.
For example, using cURL:
curl -X POST -d '{"secret": "XXX"}' URL/api/status
will return a JSON object such as:
{
"status": "success",
"cache": {
"biomodels": {
"nfiles": 8,
"size": 212254013
},
"bigg": {
"nfiles": 5,
"size": 59429817
}
},
"user": {
"uploaded": {
"nfiles": 0,
"size": 0
},
"generated": {
"nfiles": 0,
"size": 0
}
}
}
So go ahead and make sure people do not exploit your instance ;-)